Slurm job arrays¶
Job arrays let you submit many similar jobs with a single sbatch command. Each task in the array runs the same script but receives a unique index — SLURM_ARRAY_TASK_ID — which you use to vary inputs, filenames, parameters, or random seeds.
They're the right tool for embarrassingly parallel work: tasks that are fully independent, share no state, and could run in any order. Common examples include parameter sweeps, bootstrap resampling, per-sample processing pipelines, and simulation replicates.
Array size limit
The maximum number of tasks in a single array is 60000. If your workload exceeds this, contact the KIR Research Computing Team.
Basic syntax¶
This submits 10 tasks. Each receives a unique value of SLURM_ARRAY_TASK_ID from 1 to 10. You can also specify step sizes or throttle the number of concurrently running tasks:
#SBATCH --array 0-99 # 100 tasks, zero-indexed
#SBATCH --array 1-20:2 # step of 2: indices 1, 3, 5 ... 19
#SBATCH --array 1-100%5 # at most 5 tasks running simultaneously
The %N throttle is particularly useful when tasks are memory- or I/O-intensive and you want to avoid saturating the filesystem or node.

A minimal example¶
#!/bin/bash
#SBATCH --job-name array-demo
#SBATCH --time 00:10:00
#SBATCH --mem 4G
#SBATCH --array 1-5
#SBATCH --output logs/job_%a.out
#SBATCH --error logs/job_%a.err
echo "Task ${SLURM_ARRAY_TASK_ID} running on $(hostname)"
Tip
Inside #SBATCH headers, SLURM_ARRAY_TASK_ID is not yet available as a shell variable. Use the token %a in your --output and --error paths instead.
Using SLURM_ARRAY_TASK_ID¶
One file per task¶
The most common pattern in bioinformatics — one sample, one task:
SAMPLES=(sample_A sample_B sample_C sample_D sample_E)
SAMPLE=${SAMPLES[$SLURM_ARRAY_TASK_ID]}
echo "Processing ${SAMPLE}"
Set --array 0-4 to match bash's zero-based array indices.
If your files follow a naming convention, reference them directly:
Or glob them dynamically:
The glob expands in alphabetical order, so the task-to-file mapping is deterministic. Set --array to match the number of files.
Passing the index to R or Python¶
A clean pattern is to pass the task ID as a command-line argument and look up parameters from a config file:
library(optparse)
option_list <- list(
make_option("--task", type = "integer")
)
args <- parse_args(OptionParser(option_list = option_list))
params <- read.csv("params.csv")
row <- params[args$task, ]
# use row$alpha, row$beta, etc.
Or read the environment variable directly:
Setting random seeds for reproducibility¶
When running stochastic simulations, each task needs a distinct seed. Without one, tasks that start simultaneously may draw from the same pseudo-random sequence.
Avoiding conflicts between tasks¶
All tasks in an array can run simultaneously, so any shared resource — a working directory, a fixed output filename, a database connection — is a potential conflict. The fix is always the same: make everything per-task.
Give each task its own working directory when a tool writes to fixed paths you can't control:
WORKDIR=tmp/run_${SLURM_ARRAY_TASK_ID}
mkdir -p "$WORKDIR"
cd "$WORKDIR"
bash ../job.sh
mv output.tsv ../../results/output_${SLURM_ARRAY_TASK_ID}.tsv
cd ../..
rm -rf "$WORKDIR"
Use per-task paths directly when your tool accepts them:
python run.py \
--input data/sample_${SLURM_ARRAY_TASK_ID}.csv \
--output results/sample_${SLURM_ARRAY_TASK_ID}.csv
Multidimensional parameter sweeps¶
To sweep across two independent axes, encode both into a single array index using integer division and modulo:
#!/bin/bash
#SBATCH --array 0-11 # 3 models × 4 lambda values
MODELS=("lasso" "ridge" "elastic")
LAMBDAS=(0.01 0.1 1.0 10.0)
N_LAMBDA=${#LAMBDAS[@]}
MODEL=${MODELS[$(( SLURM_ARRAY_TASK_ID / N_LAMBDA ))]}
LAMBDA=${LAMBDAS[$(( SLURM_ARRAY_TASK_ID % N_LAMBDA ))]}
echo "Model: ${MODEL}, Lambda: ${LAMBDA}"
python train.py --model "$MODEL" --lambda "$LAMBDA"
```train.py --model "$MODEL" --lambda "$LAMBDA"
This produces all 12 combinations from a single array of 0–11.
Monitoring array jobs¶
Individual tasks appear in the queue as <jobid>_<taskid>.
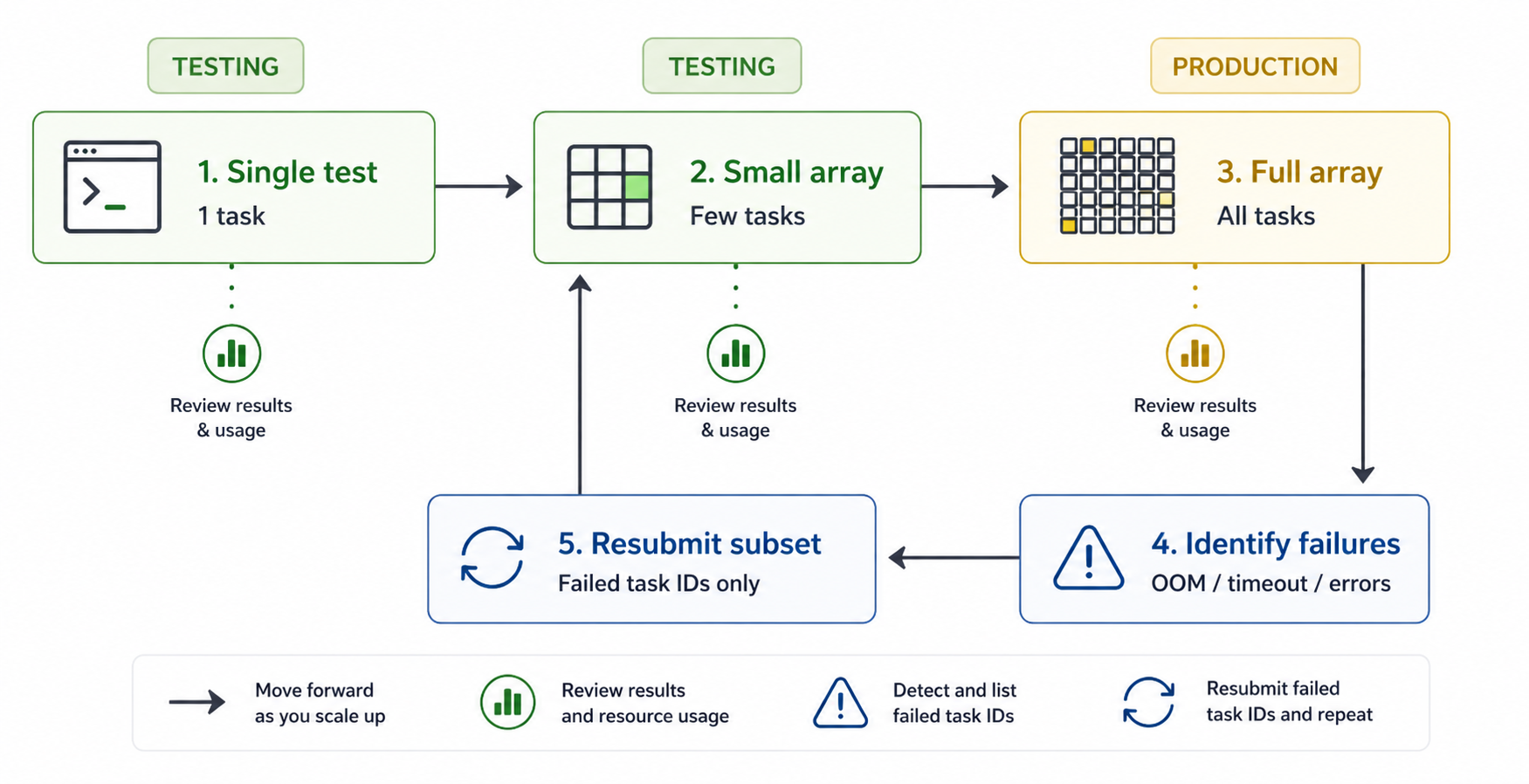
Resubmitting failed tasks¶
After an array job completes, some tasks may have failed due to timeouts, memory limits, or other transient issues. To extract the indices of all failed tasks:
sacct -j <JOBID> -n -o jobid%30,state%20 \
| awk '!/\./ && /FAILED|TIMEOUT|OUT_OF_MEMORY/ {
split($1,a,"_"); if(a[2]!="") ids=ids (ids?",":"") a[2]
} END{print ids}'
This prints a comma-separated list of task indices, e.g. 3,7,42, filtering out
job-step lines (those containing .) and catching FAILED, TIMEOUT, and
OUT_OF_MEMORY states.
Pass the output directly to --array to resubmit only the failed tasks:
As long as your script derives all input and output paths from
SLURM_ARRAY_TASK_IDand you haven't modified the directory structure, the resubmitted tasks will pick up exactly where the failed ones left off.
Combining the two steps
[[ -n "$FAILED" ]] prevents an accidental full resubmission if there
are no failed tasks.